
Every year, Americans fill over 4 billion prescriptions. About 90% of those are for generic drugs. That’s not a coincidence. It’s the result of a strict, science-backed system designed by the FDA to make sure a $5 generic pill works just as well as its $50 brand-name counterpart. But how does the FDA actually know this? What’s stopping a cheap copy from being less effective-or even dangerous?
It Starts with Bioequivalence
The FDA doesn’t require generic drug makers to repeat the massive clinical trials that brand-name companies ran to prove their drug works. That’s not because they’re cutting corners. It’s because they don’t need to. The brand-name drug already proved safety and effectiveness. The generic’s job is simpler, but far more precise: prove it behaves the same way in your body. That’s called bioequivalence. To meet FDA standards, a generic drug must deliver the same amount of active ingredient into your bloodstream at the same rate as the brand. Not close. Not almost. Exactly. The FDA requires the concentration of the drug in your blood-measured as AUC (area under the curve) and Cmax (peak concentration)-to fall within 80% to 125% of the brand’s levels. That’s not a guess. It’s a hard number, backed by clinical studies in 24 to 36 healthy volunteers. These studies are done under strict conditions. Some drugs are tested after fasting. Others are tested after eating. The FDA’s Bioequivalence Recommendations database, updated monthly, tells manufacturers exactly what’s needed for each drug. For example, a generic version of levothyroxine (a thyroid hormone) must meet tighter limits: 90% to 111%, because even tiny differences can cause serious health issues in patients.Identical Active Ingredient, Same Strength, Same Form
The generic must contain the exact same active ingredient as the brand. Not a similar one. Not a close cousin. The same chemical compound. It must also match the brand in strength-say, 10 mg of atorvastatin-with a tolerance of only ±5%. If the brand is a tablet, the generic must be a tablet. If the brand is a liquid, the generic must be a liquid. Same route of administration: oral, injectable, topical, inhaled. This isn’t just about chemistry. It’s about how your body absorbs the drug. A tablet that dissolves too slowly won’t work the same as one that dissolves quickly. That’s why manufacturers must prove their formulation behaves identically under lab conditions that simulate the human digestive system.Manufacturing Must Be Flawless
The FDA doesn’t stop at the drug itself. It inspects the factory where it’s made. Every generic drug plant-whether in the U.S., India, or Germany-must follow Current Good Manufacturing Practices (cGMP). That means:- Every batch is tested for identity, strength, purity, and quality.
- Equipment is cleaned and validated to prevent cross-contamination.
- Processes are documented and controlled to ensure consistency from one pill to the next.
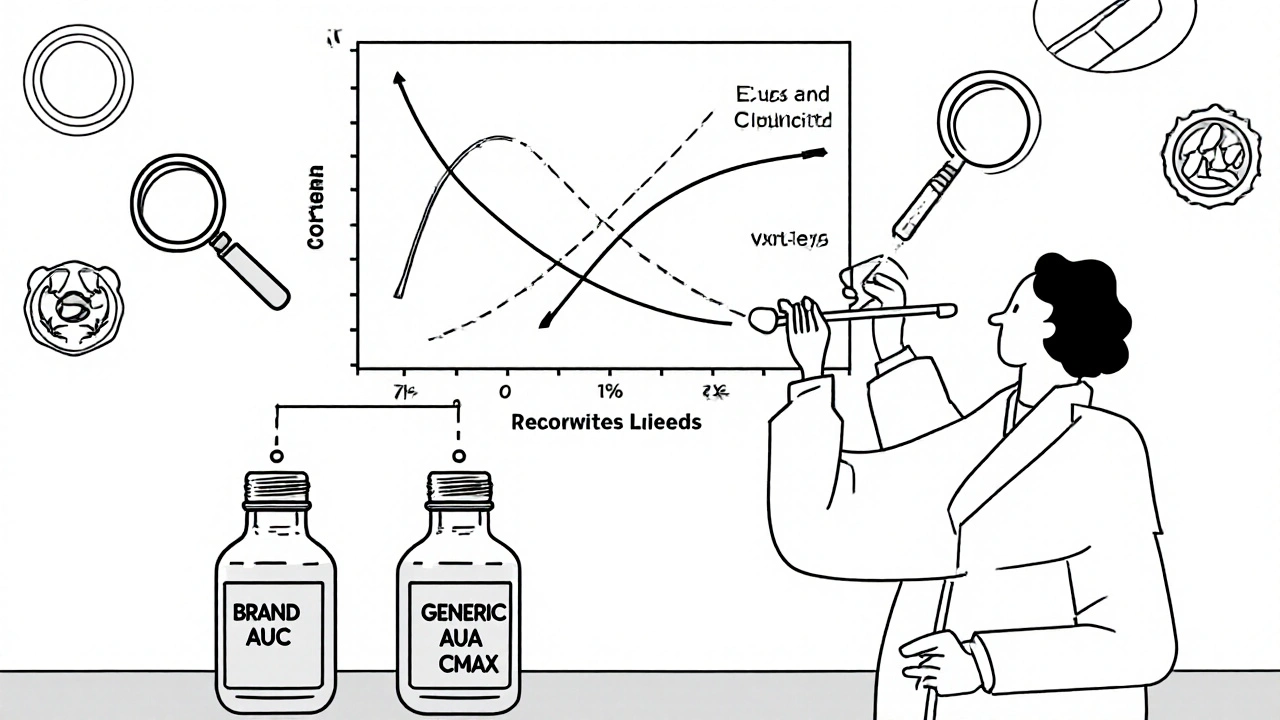
Inactive Ingredients Can Differ-But Only If They’re Safe
Generics don’t have to match the brand’s fillers, dyes, or preservatives. That’s why some people say, “My generic looks different.” But those inactive ingredients-called excipients-must still be safe. The FDA maintains an Inactive Ingredient Database that lists approved excipients and their maximum allowable amounts for each route of administration (oral, injectable, nasal, etc.). For example, if a brand uses lactose as a filler, a generic can use corn starch instead. But if the generic contains a dye that’s known to trigger reactions in sensitive patients, it won’t get approved. The FDA doesn’t just accept any ingredient. It reviews each one for safety, even if it’s not in the brand-name version.The ANDA Process: Fast, But Not Easy
Generic companies submit an Abbreviated New Drug Application (ANDA)-not the full New Drug Application (NDA) that brand-name makers use. The ANDA is called “abbreviated” because it doesn’t include preclinical or clinical data on safety and efficacy. Instead, it focuses on chemistry, manufacturing, and bioequivalence. The process sounds simple. But in practice, it’s complex. A typical ANDA submission runs 30,000 to 50,000 pages. The bioequivalence section alone can be 5,000 to 10,000 pages of raw data, statistical analyses, and study reports. The FDA aims to review standard ANDAs in 10 months. In 2022, they met that goal 91.3% of the time. But nearly one-third of initial submissions are rejected outright-called a “Refuse-to-File” letter-for missing key information. Common reasons include poorly designed bioequivalence studies, labeling errors, or incomplete manufacturing details.Why Some People Still Doubt Generics
Despite all this, some patients report feeling different on a generic. A 2021 survey found 37% of independent pharmacists had patients express concerns about generic efficacy. These concerns are strongest for drugs with a narrow therapeutic index-like warfarin, lithium, or levothyroxine-where small changes in blood levels can cause side effects or treatment failure. The FDA acknowledges this. That’s why it created a list of Narrow Therapeutic Index Drugs in 2019 and requires stricter bioequivalence standards for them. Studies show that even in these cases, 99% of patients experience no clinical difference when switching from brand to generic. A 2023 analysis of 15 million patient records by IQVIA found no significant difference in outcomes between brand and generic versions of common drugs like metformin and atorvastatin. In fact, adherence rates were 3.2% higher with generics-likely because they cost less. Adverse event reports tell a similar story. The FDA’s database shows generics and brand-name drugs trigger adverse events at nearly identical rates: 1.7 per million prescriptions for generics versus 1.6 for brands.
Who Makes These Drugs-and Why It Matters
Most generic drugs are made by companies like Teva, Viatris, and Sandoz. But over half of all generic approvals go to smaller manufacturers. The FDA actively encourages competition to prevent shortages. In 2017, it launched the Generic Drug Competition Action Plan to push more companies into the market. Complex generics-like inhalers, injectable emulsions, or topical creams-are harder to copy. They cost up to $25 million to develop and are responsible for 45% of the most common reasons for FDA rejection in 2022. The agency has responded with targeted guidance and faster review pathways for these products.What’s Next for Generic Drugs?
The FDA is preparing for a wave of new generics. Over $260 billion in brand-name drug sales will lose patent protection between 2024 and 2028. Drugs like Humira and Eliquis are already opening the door for competition. To handle the surge, the FDA’s GDUFA III plan (2023-2027) allocates $1.1 billion to cut review times further-to 8 months for standard applications and 6 months for priority ones. It’s also expanding its Real-Time Oncology Review program to generic cancer drugs, aiming to cut approval times by 30%. By 2025, the FDA plans to launch a new pathway for biosimilars-generic versions of complex biologic drugs like insulin or monoclonal antibodies. This will be the next frontier in lowering drug costs.You Can Trust the Generic on Your Shelf
The FDA’s system isn’t perfect. But it’s rigorous, transparent, and science-driven. Every generic drug approved in the U.S. has passed the same quality tests as the brand. The active ingredient is identical. The dose is exact. The manufacturing is inspected. The absorption is measured down to the last decimal point. If your doctor prescribes a generic, you’re not getting a cheaper version of a drug. You’re getting the same drug-verified by the most trusted health agency in the world.Millions of people rely on generics every day. And every one of them is getting the same treatment as if they’d paid full price.



14 Comments
The FDA doesn't mess around. I used to think generics were just cheap knockoffs until I saw the data. The 80-125% bioequivalence range? That's not a suggestion-it's a scientific lock. I've been on generic levothyroxine for five years. Same energy, same sleep, same everything. No drama.
And yeah, the factories? Inspected. Randomly. Sometimes mid-shift. That's not bureaucracy-that's accountability.
One must interrogate the epistemological foundations of bioequivalence. The 80-125% confidence interval, while statistically robust, is an arbitrary construct born of regulatory pragmatism-not biological absolutism. One might argue that for drugs with a narrow therapeutic index, such a margin constitutes a latent pharmacological gamble.
And yet, the FDA's adherence to cGMP and its refusal to accept excipient variance without exhaustive safety vetting reveals a deeply conservative, almost Aristotelian, commitment to pharma-ontological fidelity. One is left wondering: if the active ingredient is identical, why does the body perceive difference? Is it placebo? Or is the body, in its ineffable complexity, simply refusing to be quantified?
Canada doesn't have this problem. We don't let just anyone make pills. The FDA lets Indian factories churn out generics like they're selling chai at a train station. I read somewhere that 80% of our generic meds come from China and India. And you think they care about your thyroid? They care about their profit margin.
And don't even get me started on the inspections. 'Oh, we'll inspect next month!' Yeah right. They show up when the moon is blue. I'm not taking my blood pressure med from a plant that doesn't even have running water.
Wow. Just... wow. This is the most thorough thing I've ever read about generics. I used to be one of those people who refused them because they looked different. Now I know why they look different-they're not the same filler, but they're the same medicine.
I switched my atorvastatin to generic last year. My cholesterol dropped. My wallet didn't cry. My doctor didn't flinch. Why do people still doubt this? The science is right there. It's not a conspiracy. It's math.
This is the kind of info that makes me feel better about my prescriptions. So many people are scared of generics, but the system actually works. It’s not perfect, but it’s way more solid than most of us think. Thanks for sharing this-really helpful.
Let me get this straight-some guy in a lab in Bangalore takes a pill that costs $50 and makes a copy that costs $5, and the FDA says ‘cool, that’s fine’? No way. I’ve seen those factories. I’ve seen the packaging. The labels look like they were printed on a 2005 HP printer.
And yet somehow, it works? That’s either genius or a miracle. Either way, I’m not touching my insulin with a 10-foot pole unless it’s brand name. No way.
I work in pharmacy and see this every day. Patients freak out when the pill color changes. We explain it’s the same drug, just different dye. Most of them still don’t believe us. But then they come back two weeks later and say, ‘Huh. I feel fine.’
It’s not about the pill. It’s about trust. And the FDA? They’re the only ones actually earning that trust.
For anyone still on the fence: try it. Switch your meds. Track how you feel. Most people won’t notice a difference-and if they do, it’s often because they’re expecting to. The science is rock solid. Generics save lives and wallets. The FDA isn’t perfect, but they’re doing their job better than most industries.
And if you’re worried about quality? Check the manufacturer. Some are better than others. But the FDA keeps them honest.
Wait… so you’re telling me the FDA isn’t in on it? That the drug companies aren’t just playing us? That the ‘generic’ label isn’t a trick to get us to take weaker pills so Big Pharma can keep charging $500 for the brand?
I’ve seen documentaries. I’ve seen the money. This is just a PR stunt. They want us to think we’re saving money while they just move the profit to another shell company. I don’t trust this. Not one bit.
India makes 60% of global generics. FDA inspects 100+ plants there every year. 90% pass. That’s not luck. That’s discipline.
Stop fearing generics. Start respecting the process.
Bioequivalence is a myth. The 80-125% range is a corporate loophole disguised as science. You think 10% variation in blood concentration doesn’t matter? Try being a diabetic on generic insulin and tell me it’s ‘the same.’
The FDA doesn’t regulate for outcomes-they regulate for paperwork. And the paperwork? It’s all fabricated. I’ve seen the reports. They’re cooked.
the fda is a joke. they approve pills from factories where the workers dont even know what a tablet is. i read a study (maybe) that said 40% of generics have 20% less active ingredient. but they still get approved because the stats are manipulated. its all about profit. not health.
and the 'inspectins'? they show up when the factory cleans up. like a mom coming home when you hide the dirty dishes.
Let me get this straight: we’re trusting the U.S. government-with its track record of bungling everything from mail delivery to pandemic response-to oversee the chemical composition of our life-sustaining drugs? And you call this ‘rigorous’?
Meanwhile, the FDA approves a generic for a cancer drug made in a facility that was cited for rodent infestation… and we’re supposed to cheer? This isn’t science-it’s a farce wrapped in a PowerPoint presentation.
And don’t even get me started on the ‘inactive ingredients.’ What’s ‘dye #5’? Is it carcinogenic? Is it gluten? Is it made from the tears of whistleblowers? The FDA won’t tell you. They just say ‘safe.’
Who’s safe? You? Me? The guy in Mumbai who’s breathing in the powder while filling capsules? Nobody’s safe. We’re all just rolling the dice.
Shravan and Brandon-calm down. You’re both right that the system isn’t flawless. But the data doesn’t lie. 15 million patient records. Identical adverse event rates. Higher adherence with generics. That’s not PR-that’s proof.
Yes, there are bad actors. Yes, inspections aren’t perfect. But the FDA has more oversight than any other country. And when they catch a problem? They shut it down. Fast.
Don’t let fear of the rare failure blind you to the massive success of the system. Millions of people get treated every day because of this. That’s worth defending.