
When a new drug hits the market, the work isn’t done. In fact, the real safety testing often begins only after patients start using it daily - not in controlled clinical trials with 5,000 people, but in millions of real lives with different ages, health conditions, and medications. Tracking post-marketing studies for drug safety isn’t optional. It’s the backbone of protecting public health. And if you’re responsible for managing this process - whether you’re in pharma, regulatory affairs, or clinical research - you need a clear, practical system to stay ahead of hidden risks.
Why Post-Marketing Surveillance Matters
Pre-approval clinical trials are designed to prove a drug works and isn’t immediately dangerous. But they can’t catch everything. Rare side effects? They might show up in 1 in 10,000 patients. That’s invisible in a trial of 3,000 people. What about interactions with other drugs elderly patients take? Or effects on pregnant women, who were often excluded from trials? These gaps aren’t flaws - they’re limitations of science under time and cost pressure. That’s where post-marketing surveillance (PMS) comes in. It’s the ongoing watch after approval. The U.S. FDA, Health Canada, the UK’s Yellow Card system, and the EU’s EudraVigilance all rely on it. In 2022, over 76,000 adverse reaction reports came in through the UK’s system alone. In the U.S., the FDA’s FAERS database held more than 30 million reports. These aren’t just numbers. Each one could be a clue to a serious, previously unknown risk.The Three-Step Framework for Tracking Studies
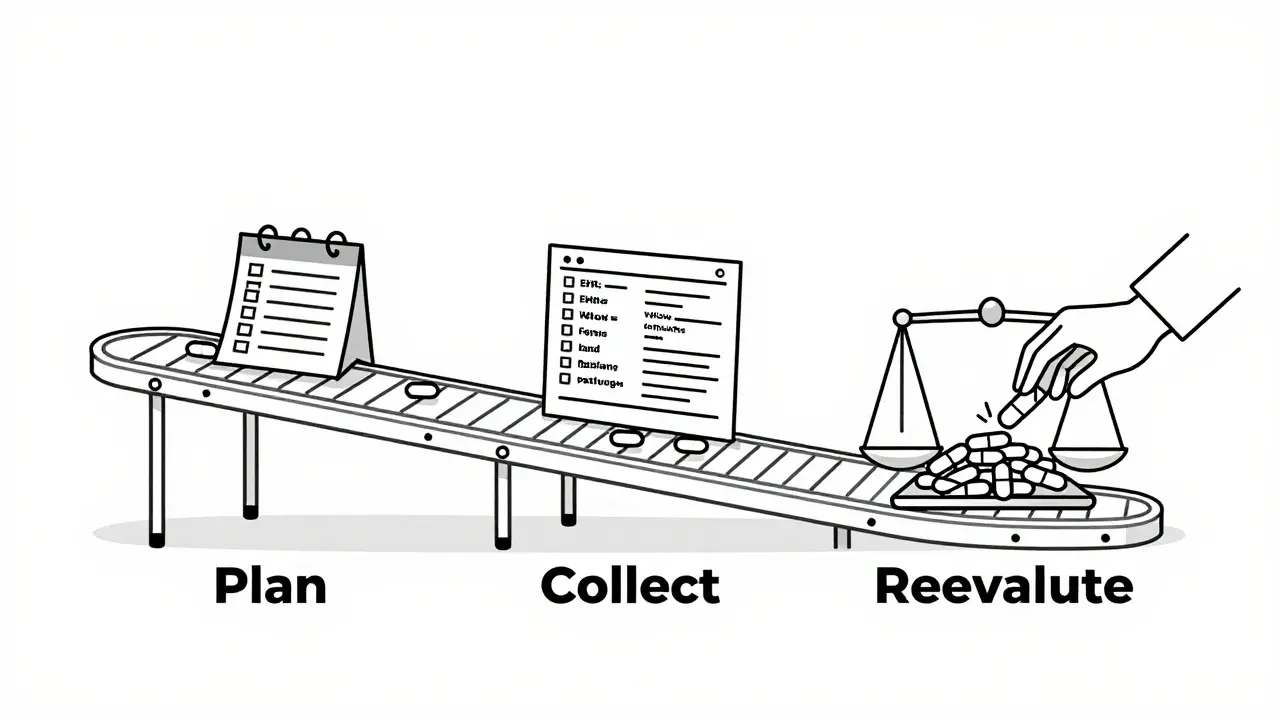
You can’t track everything at once. You need structure. The industry standard breaks down into three phases:- Plan your surveillance - Before the drug launches, you must have a Safety Surveillance Plan (SSP) and a Risk Minimization Plan (RMP). The SSP outlines how you’ll collect data: spontaneous reports, electronic health records, patient registries. The RMP details how you’ll reduce harm - like special patient guides, restricted distribution, or mandatory training for prescribers.
- Collect and report - After launch, you’re required to submit periodic safety updates. These include spontaneous reports from doctors and patients, results from dedicated post-marketing studies, and data pulled from real-world sources like insurance claims or hospital records. The FDA mandates these reports quarterly at first, then annually.
- Reevaluate every 4-10 years - Regulators don’t just check in once. Companies must formally reconfirm the drug’s safety, quality, and effectiveness within a set window. If new data shows risks outweigh benefits, the label changes, or the drug gets pulled.
Tools You Can’t Ignore: FAERS and Sentinel
Two systems dominate U.S. safety monitoring: FAERS and Sentinel. They’re not the same - and you need both.FAERS (FDA Adverse Event Reporting System) is the oldest and most widely used. It’s a database where anyone - doctors, patients, pharmacists, or the drug maker - can submit reports of side effects. It’s passive. It relies on people noticing and reporting. But it’s powerful because it’s broad. In 2022, 63% of safety actions by the FDA were triggered by FAERS reports. That’s the frontline.
Sentinel is active surveillance. It pulls data from over 300 million Americans - insurance claims, electronic health records, pharmacy logs. It doesn’t wait for reports. It searches for patterns. Did patients on Drug X have a spike in liver failure three months after starting it? Sentinel finds that. In 2023, the system expanded to include detailed EHR data from 24 million people across six partners. That’s a game-changer. It can see lab results, vital signs, diagnoses - things FAERS can’t.
But here’s the catch: Sentinel still can’t replace FAERS. It’s great for trends, but it misses rare events. FAERS catches the outliers. Together, they cover more ground than either alone.
Global Systems and How They Compare
The U.S. isn’t alone. Other countries run similar systems - but differently.- UK (Yellow Card): Simple, public-facing. Anyone can report. In 2022, submissions rose 12% year-over-year. It’s easy to use, which drives participation.
- Canada (Canada Vigilance): Similar to FAERS, but with tighter integration into provincial health systems. Received nearly 29,000 reports in 2022.
- EU (EudraVigilance): Centralized across all member states. Now adding AI to detect signals faster. Rolling out in 2025.
Each system has strengths. The UK’s simplicity encourages public reporting. Canada’s integration with healthcare providers improves data quality. The EU’s scale allows for cross-border signal detection. If you’re managing a global drug, you need to track all of them - not just the U.S. data.
How Signals Turn Into Action
Finding a signal is just the start. What happens next matters.The FDA uses a five-phase process:
- Identify - A pattern emerges in FAERS or Sentinel. Maybe 15 cases of sudden kidney failure in patients on a new diabetes drug.
- Triage - Is this a one-off? Or a trend? Teams prioritize based on severity and number of cases.
- Evaluate - Epidemiologists, statisticians, and clinicians dig into data. They check if it’s the drug, or something else - like patients also taking a different medication.
- Act - If the signal holds, the FDA issues a Drug Safety Communication. They may update the label, send a letter to doctors, or require a Risk Evaluation and Mitigation Strategy (REMS).
- Communicate - Findings go public. The FDA posts updates quarterly. Drug makers must notify prescribers.
Between 2018 and 2022, 87% of safety actions led to label changes. Only 3% led to REMS updates. Less than 1% resulted in withdrawal. Most risks are manageable - if caught early.
Why So Many Studies Run Late
The FDA can require companies to run post-marketing studies. But here’s the problem: most run late.A 2023 National Academies report found that 72% of mandated studies missed their deadlines. The average completion time? 5.3 years - over two years past the 3-year requirement. Why?
- Recruiting patients is hard. Many trials need specific health profiles.
- Data collection across hospitals and clinics is fragmented.
- Companies don’t always assign enough staff or budget.
Companies that succeed use distributed data networks - systems that connect multiple healthcare providers through secure, standardized data formats. These cut study setup time from 14 months in 2018 to under 9 months in 2023. If you’re managing a study, don’t wait until launch to build your data pipeline. Start before the drug even hits shelves.
Emerging Tech: AI, LLMs, and the Future
Artificial intelligence is changing how we spot danger. The FDA and Lifebit AI ran a pilot in 2023 using Large Language Models (LLMs) to scan unstructured EHR notes - doctor’s handwritten observations, discharge summaries, progress notes.Result? Signal detection improved by 42%. But false positives went up 23%. That’s the trade-off. AI finds things humans miss - but it also sees patterns that aren’t real. You still need expert review.
Future systems will combine AI with genomic data. The FDA’s Sentinel Common Data Model Plus (SCDM+) will integrate DNA data with clinical records for 50 million patients by 2026. Imagine knowing a patient’s genetic risk for liver toxicity before prescribing a drug. That’s the next level.
By 2027, the WHO plans to link pharmacovigilance data across 100 countries. That means a rare side effect spotted in Japan could be flagged in Brazil within days - not years.
Best Practices for Effective Tracking
If you’re building or managing a post-marketing surveillance system, here’s what works:- Assign dedicated pharmacovigilance staff - Aim for one specialist per $500 million in annual drug revenue. Too few people? Reports pile up. Alerts get missed.
- Use automated alerts - Set up triggers for unusual spikes in adverse reports. Don’t wait for monthly reports.
- Track your timeliness - Measure the Post-Marketing Study Timeliness Index (PMSTI): what % of studies finish on time? If it’s below 70%, you’ve got a problem.
- Integrate global data - Don’t just look at U.S. reports. Connect to EudraVigilance and Canada Vigilance. Safety is global.
- Test your systems - Run mock signal detection exercises quarterly. Can your team respond in 48 hours?
What Happens If You Don’t Track Properly?
Ignoring post-marketing surveillance isn’t just risky - it’s costly. The FDA can issue warning letters, halt sales, or demand recalls. In 2020, a popular painkiller was pulled after post-marketing data linked it to rare but fatal liver damage. The company had ignored early signals for over two years.Reputation damage? That’s harder to measure. But patients lose trust. Doctors stop prescribing. Revenue drops. And regulators watch you closer for the next 10 years.
Post-marketing surveillance isn’t a box to check. It’s a continuous conversation between science, data, and real-world use. The drug doesn’t stop being monitored after approval - it starts being truly tested.
What’s the difference between spontaneous reporting and active surveillance?
Spontaneous reporting means someone - a doctor, patient, or pharmacist - notices a side effect and reports it voluntarily. That’s how FAERS works. Active surveillance means systems automatically scan large datasets (like insurance claims or EHRs) to find patterns without waiting for a report. Sentinel is an example. Spontaneous reports catch rare events; active surveillance finds trends faster.
How long do post-marketing studies usually take to complete?
Regulators often require completion within 3 years, but in reality, most take over 5 years. A 2023 National Academies report found 72% of studies missed deadlines. Delays happen because recruiting patients is slow, data systems aren’t connected, and companies don’t prioritize these studies early enough.
Can AI replace human reviewers in drug safety monitoring?
No. AI can speed up signal detection - FDA pilots showed 42% better accuracy when analyzing EHR notes. But false positives increased by 23%. AI finds patterns humans miss, but it also creates noise. Human experts are still needed to confirm whether a signal is real, assess risk, and decide on action.
What’s the most common safety action taken after a post-marketing signal?
Updating the drug label. Between 2018 and 2022, 87% of safety actions led to changes in the prescribing information - like adding new warnings, contraindications, or dosage adjustments. Letters to doctors and REMS modifications happen less often. Withdrawals are rare - under 1%.
Why are elderly patients underrepresented in clinical trials but critical in post-marketing?
Clinical trials often exclude older adults due to complex health conditions or multiple medications. But they’re the biggest users of prescription drugs. In the EU, 43% of actual users were over 65 - yet only 15% of trial participants were. That’s why 28% of serious side effects detected after launch wouldn’t have been found in trials. Post-marketing data fills this gap.
What’s the best way to improve post-marketing study timelines?
Use distributed data networks that connect hospitals, clinics, and pharmacies through standardized formats. These cut study setup time from 14 months to under 9 months. Also, assign a dedicated team early - don’t wait until after launch. Start building data access agreements and patient recruitment plans during clinical trial phases.

14 Comments
I've seen too many drugs get approved with zero real-world data. Then people start dying and suddenly everyone's shocked. This post nails it - safety doesn't end at FDA approval. It just begins.
Real talk - if your company treats post-market surveillance like a box to tick, you’re already failing. Patients aren’t lab rats. They’re moms, dads, grandpas with three meds in their pill organizer. We owe them better.
The distinction between FAERS and Sentinel is critical. Passive reporting captures outliers; active surveillance detects trends. Both are necessary, not interchangeable. This framework is sound.
Yo, AI is gonna change everything but let’s be real - it’s still a hype train. I’ve seen LLMs flag ‘headache’ as a signal 200 times in a week because some doc wrote ‘pt had a bad headache’ in notes. Still need humans to filter the noise.
Yankees think they’ve got the best system? EudraVigilance handles 28 countries with one database. We’ve been doing this properly since the 90s. FAERS is a spreadsheet with a fancy name.
I work in a small clinic in Kerala and we report everything we see - even if it’s just a weird rash or dizziness. I know it feels tiny, but those reports? They matter. One time we flagged a combo drug that later got pulled in the UK. We didn’t know we were part of something bigger until the email came. It gave me chills. Don’t underestimate the power of small actions.
This post is just pharma propaganda. They want you to think they care. But they delay studies, bury data, and only act when the media blows up. Wake up. This whole system is rigged.
I must say, the structural integrity of this exposition is... profoundly underwhelming. The syntactic architecture lacks the requisite rigor one would expect from a discourse on pharmacovigilance. One wonders if the author has ever read a peer-reviewed journal.
You’re not just tracking drugs - you’re tracking people. Every report is someone’s story. A grandma. A kid. A veteran. Treat it like that. And hey - if you’re reading this and you’re in pharma? You’re doing God’s work. Keep going. 🙏
My aunt took that new painkiller and got sick. We reported it. No one called back. But it’s still in the system. I hope it helps someone else.
I know the truth. The FDA and Big Pharma are in bed together. They let dangerous drugs through on purpose so they can sell more pills later. The ‘safety actions’? Just theater. Watch the money. It always follows the cover-up.
Honestly, this is the most coherent piece on pharmacovigilance I’ve read in years. No fluff. Just facts. And the part about elderly patients? Spot on. They’re the most vulnerable and the most ignored. Thank you.
The integration of genomic data into the Sentinel Common Data Model Plus represents a paradigm shift in pharmacovigilance. By 2026, with 50 million patients’ genomic profiles linked to longitudinal clinical outcomes, we will transition from reactive surveillance to predictive safety architecture. This is not merely innovation - it is the evolution of ethical medicine.
You say ‘72% of studies run late’? That’s because the FDA doesn’t even enforce deadlines. Meanwhile, we’re wasting billions on AI that can’t tell a migraine from a stroke. This whole system is a joke. Fix the basics before you throw in fancy tech.