Generic drugs make up about 90% of all prescriptions filled in the United States. You likely take one right now. But here is the thing that often gets overlooked: unlike brand-name drugs, which undergo massive clinical trials before hitting shelves, generics rely on a shortcut called bioequivalence to get approved. They just need to prove they act like the original drug in your body. So, once they are out there, how does the Food and Drug Administration (FDA) actually keep an eye on them? The answer lies in a complex web of data mining, patient reports, and increasingly, artificial intelligence.
The short version is that the FDA doesn't just approve a generic and walk away. They use a system called post-market surveillance, which is essentially continuous monitoring of drug safety in the real world. Because clinical trials only involve a few thousand people, they can't catch every rare side effect or manufacturing glitch. Post-market surveillance catches what the trials miss. For generics, this is even trickier because multiple manufacturers produce the same drug, making it hard to pinpoint exactly which batch caused a problem.
The Foundation: Bioequivalence and the Hatch-Waxman Act
To understand why surveillance is so critical for generics, you first have to understand how they get approved. In 1984, Congress passed the Hatch-Waxman Act. This law was designed to balance two things: encouraging new drug innovation while also allowing cheaper generic versions to enter the market quickly. Under this act, a generic sponsor doesn't need to run expensive, years-long clinical trials to prove their drug works. Instead, they submit an Abbreviated New Drug Application (ANDA).
The key requirement here is bioequivalence. This means the generic must deliver the same amount of active ingredient into the bloodstream at the same rate as the reference listed drug (the brand name). If the math checks out, the FDA assumes the therapeutic effect will be the same. However, "same" isn't always perfect, especially for complex generics. These are drugs that are harder to manufacture, like inhalers, topical creams, or extended-release pills. For these, small differences in inactive ingredients or delivery mechanisms might not show up in a simple blood test but could still affect how well the drug works in practice. That is where post-market surveillance steps in.
The Data Engines: FAERS and MedWatch
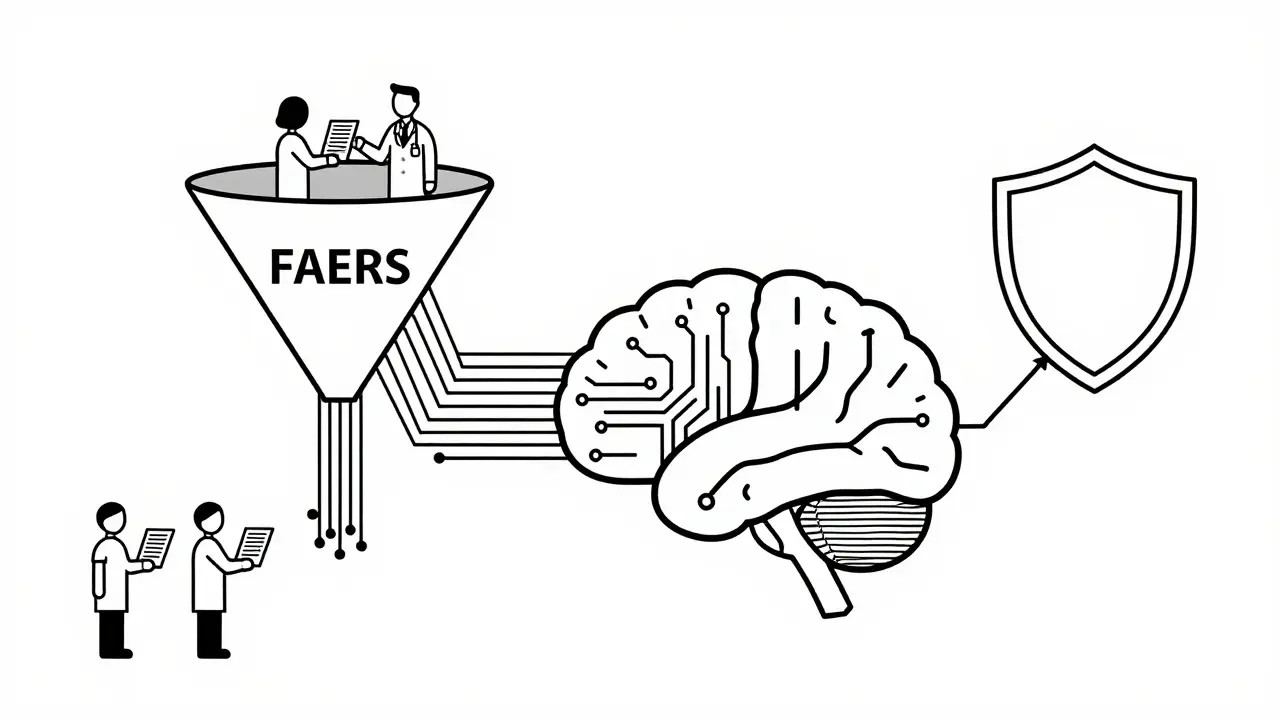
The backbone of FDA's generic monitoring is the FDA Adverse Event Reporting System (FAERS). Think of FAERS as a giant digital bucket where everyone dumps safety concerns. Healthcare professionals, patients, and consumers can report unexpected side effects, medication errors, or product quality issues through a program called MedWatch. Once submitted, these reports flow into FAERS.
Here is how it works in practice. If a nurse notices a patient having severe rashes after switching from Brand X Generic A to Brand Y Generic B, she can file a report. The FDA’s Center for Drug Evaluation and Research (CDER), specifically the Office of Surveillance and Epidemiology, then analyzes these reports. They look for "signals." A signal is basically a statistical anomaly-a specific side effect appearing more frequently than expected for a specific drug. It is important to remember that a report alone does not prove the drug caused the issue. It just flags something for deeper investigation. The FDA uses sophisticated algorithms to sift through millions of these reports to find patterns that human reviewers might miss.
The Sentinel Initiative: Proactive Monitoring
Relying solely on people to report problems has a big flaw: underreporting. Most people never report side effects unless they are life-threatening. To fix this, the FDA launched the Sentinel Initiative in 2008. Unlike FAERS, which is reactive, Sentinel is proactive. It taps into existing electronic health databases from insurance claims, electronic health records (EHRs), and registries.
As of 2023, the Sentinel System covers over 200 million Americans. This allows the FDA to run queries across vast populations without waiting for individual reports. For example, if there is a theoretical concern that a certain generic statin might increase liver enzyme levels, the FDA can query the Sentinel database to see if patients taking that specific generic show higher rates of liver issues compared to those taking other statins. This shift from passive reporting to active data mining has been a game-changer for detecting subtle safety signals in generic drugs, particularly when comparing different manufacturers' versions of the same molecule.
The Challenge of Complex Generics
Not all generics are created equal. Simple tablets that dissolve instantly are relatively easy to monitor because bioequivalence is a strong predictor of performance. But complex generics pose a unique headache for regulators. These include products like insulin injections, ophthalmic solutions, and inhalers. For an inhaler, for instance, the particle size and how deeply the drug penetrates the lungs matter just as much as the chemical composition. Standard bioequivalence studies might not fully capture these nuances.
The National Academies of Sciences, Engineering, and Medicine highlighted this gap in a 2021 report, noting that current surveillance methods struggle to detect therapeutic inequivalence in complex generics. Critics argue that the FDA lacks sufficient resources to track these specific formulation differences. When a patient switches from a brand-name inhaler to a generic, and their asthma control worsens, it is difficult to know if it was the device mechanics, the powder formulation, or just patient perception. This ambiguity makes post-market surveillance for complex generics a high-stakes, low-visibility area of regulatory science.
Manufacturing Inspections and Quality Control
Safety isn't just about side effects; it is also about manufacturing consistency. The FDA conducts periodic, unannounced inspections of drug production facilities. Field investigators check whether companies are adhering to Current Good Manufacturing Practices (CGMP). For generics, this is crucial because slight variations in the manufacturing process-like temperature changes during mixing or impurities in raw materials-can lead to inconsistent dosing.
If the FDA finds violations, they can take enforcement actions. This might range from warning letters to mandatory recalls. In recent years, there has been increased scrutiny on foreign manufacturing sites, where many generic APIs (Active Pharmaceutical Ingredients) are produced. Ensuring that the generic pill you buy at the pharmacy contains exactly what the label says is a core part of post-market surveillance, separate from pharmacovigilance (side effect tracking).
The Role of AI and Machine Learning
The volume of data coming from FAERS and Sentinel is overwhelming. Traditional epidemiological methods are slow and labor-intensive. Recognizing this, the FDA has begun integrating artificial intelligence (AI) and machine learning (ML) into its surveillance toolkit. In fiscal year 2023, the agency allocated $5.2 million specifically for AI/ML-based postmarket surveillance research.
The goal is to automate signal detection. Imagine an algorithm that can scan millions of social media posts, medical forum discussions, and EHR notes simultaneously to identify emerging complaints about a specific generic drug weeks before they appear in formal FAERS reports. Industry analysts predict that by 2027, AI-enhanced systems could reduce the time to detect safety signals for complex generics by 60-70%. This would allow the FDA to intervene faster, potentially preventing widespread adverse events. However, challenges remain in validating these AI models to ensure they don't generate false positives, which could unnecessarily alarm the public or disrupt drug supply chains.
Patient Perception and the Nocebo Effect
A significant hurdle in generic surveillance is the "nocebo effect." This occurs when patients expect a negative outcome because they distrust generics, leading them to experience or report side effects that aren't pharmacologically caused by the drug. A 2019 study in JAMA Internal Medicine found that about 15% of adverse event reports for generics involved complaints about perceived differences in efficacy compared to brand names, even though no causal relationship was established.
This creates noise in the surveillance data. The FDA has to distinguish between genuine safety signals and patient dissatisfaction driven by marketing or misinformation. Dr. Gary Buehler, former Director of the Office of Generic Drugs, noted that patient perceptions can impact acceptance and even outcomes. Navigating this psychological layer requires careful epidemiological follow-up to confirm whether a reported cluster of events is real or artifact.
Regulatory Actions and Outcomes
When the FDA identifies a confirmed safety signal in a generic drug, several actions can follow. The most common is updating the product labeling to include new warnings or contraindications. If the risk is severe, the FDA may issue a "Dear Healthcare Provider" letter to alert doctors and pharmacists. In extreme cases, such as contamination or serious manufacturing defects, the agency can mandate a voluntary recall or take legal action against the manufacturer.
For complex generics, the FDA may require additional post-marketing studies. This is less common but increasingly discussed as part of the "Best Practices in Drug and Biological Product Postmarket Safety Surveillance" guidance issued in 2019. The aim is to create a more robust framework for monitoring these tricky products throughout their lifecycle, ensuring that the convenience of lower prices doesn't come at the cost of patient safety.
How does the FDA monitor generic drugs differently from brand-name drugs?
The FDA monitors both using similar tools like FAERS and Sentinel, but the focus differs. Brand-name drugs have extensive pre-market clinical trial data. Generics rely on bioequivalence data, so post-market surveillance is critical to catch any real-world differences that weren't apparent in initial testing, especially for complex generics where formulation matters.
What is the Sentinel Initiative?
The Sentinel Initiative is a proactive surveillance system used by the FDA. It accesses large electronic healthcare databases, including insurance claims and electronic health records, covering over 200 million Americans. It allows the FDA to run queries to detect safety signals in real-time without waiting for individual adverse event reports.
Why are complex generics harder to monitor?
Complex generics, such as inhalers, topical creams, and extended-release formulations, have delivery mechanisms that are difficult to replicate exactly. Standard bioequivalence tests measure blood concentration but may not capture differences in how the drug is delivered to the target site (e.g., lung depth for inhalers). This makes it harder to ensure therapeutic equivalence in the real world.
Can I report a side effect from a generic drug?
Yes. You can report side effects, medication errors, or quality issues through the FDA's MedWatch program. These reports go into the FAERS database, where safety evaluators analyze them for potential signals. Your report helps the FDA identify problems that might not be seen in clinical trials.
Is the FDA using AI to monitor drug safety?
Yes, the FDA is actively investing in artificial intelligence and machine learning to enhance post-market surveillance. The goal is to automate the detection of safety signals from large datasets, reducing the time to identify risks from months to weeks, particularly for complex generic drugs.



13 Comments
I actually find this whole process fascinating, especially how they use AI to sift through millions of reports. It's pretty cool that technology can help catch things human reviewers might miss in such a massive dataset. I'm curious about the accuracy though, do you think the algorithms are getting better at distinguishing real side effects from noise?
The concept of bioequivalence is fundamentally flawed when applied to complex generics. The assumption that identical blood concentration equals identical therapeutic effect is scientifically naive. Inhalers and topical creams require precise delivery mechanisms that standard pharmacokinetic studies fail to capture adequately. This regulatory shortcut prioritizes cost over patient safety, creating a dangerous blind spot in post-market surveillance. We need rigorous clinical trials for all generic formulations, not just simple tablets.
Another day, another government agency pretending they have everything under control while importing cheap drugs from overseas factories who don't even follow basic hygiene standards. You want me to trust that some random batch from a plant I've never heard of is safe? Please. They cut corners everywhere except on their own salaries.
It is interesting to consider the philosophical implications of trusting statistical models over individual experience. When we rely on big data for health decisions, we risk losing the nuance of personal biology. Perhaps there is a middle ground where both approaches coexist harmoniously without dismissing either perspective entirely.
Oh sure, let's just blame the FDA for everything while ignoring the fact that these companies cut costs by using cheaper inactive ingredients. But hey, as long as the active ingredient is technically present, who cares if the pill doesn't dissolve properly? Just swallow your pride along with your medication.
lol yeah right 🙄 like the FDA gives a damn about us little people. They're too busy playing favorites with big pharma. Anyway, good luck trying to report a side effect unless you have a lawyer and a lot of free time 😂
i took a generic antidepressant last year and felt terrible so i switched back to brand name immediately. why would anyone trust those fake pills anyway its just greed driving this whole system and nobody seems to care until someone gets hurt
The epistemological crisis within current pharmacovigilance frameworks necessitates a paradigm shift towards more robust evidentiary standards. Relying solely on passive reporting systems like FAERS creates significant selection bias and confounding variables that render the resulting data largely inconclusive for determining causality in complex generic formulations.
maybe we should look at how other countries handle this instead of just complaining. India has strict quality controls now too and many generics exported from here meet international standards. Let's keep an open mind about global manufacturing improvements 👍
In Canada, we often see similar challenges with our generic drug approval processes, but there is a growing emphasis on patient-centered outcomes rather than just biochemical equivalence. It is wonderful to see discussions around improving surveillance methods because ultimately, what matters most is ensuring that every patient receives consistent and reliable care regardless of which manufacturer produced their medication, and fostering international cooperation could really enhance our collective ability to monitor drug safety effectively across borders.
As someone who works in healthcare, I can tell you that the nocebo effect is real and powerful. Patients expect generics to be worse, so they report more issues. But the Sentinel Initiative is genuinely helpful for catching real problems that slip through the cracks. We need to educate patients better about what bioequivalence actually means.
I totally understand the concern about complex generics like inhalers, and it makes sense that small differences in delivery could impact effectiveness significantly. It is reassuring to know that the FDA is investing in AI to help detect these subtle signals faster, and hopefully, this will lead to better support for patients who rely on these medications daily. Let's hope for continued improvements in transparency and monitoring!
Just wanted to say thanks for breaking this down so clearly! I always wondered how they track all those generic drugs out there. It's wild to think about all the data being processed behind the scenes. Keep up the great work sharing info like this! ✨