What Corrective Actions Really Mean in Manufacturing
When a factory finds a batch of defective parts, it’s tempting to throw them out, tweak a machine, and move on. But that’s not a corrective action-that’s a quick fix. True corrective action is about digging deeper. It’s asking: Why did this happen? and How do we make sure it never happens again? This isn’t just about fixing one bad batch. It’s about stopping the same mistake from repeating across hundreds or thousands of units.
Manufacturers in regulated industries like medical devices, pharmaceuticals, and aerospace don’t have the luxury of guesswork. If a heart monitor fails because of a loose wire, and that failure isn’t properly traced back to its source, lives could be at risk. That’s why the industry follows a formal system called CAPA-Corrective and Preventive Action. It’s not optional. It’s required by FDA 21 CFR Part 820, ISO 13485, and cGMP rules. And it’s not paperwork for the sake of paperwork. It’s the difference between a company that survives inspections and one that gets shut down.
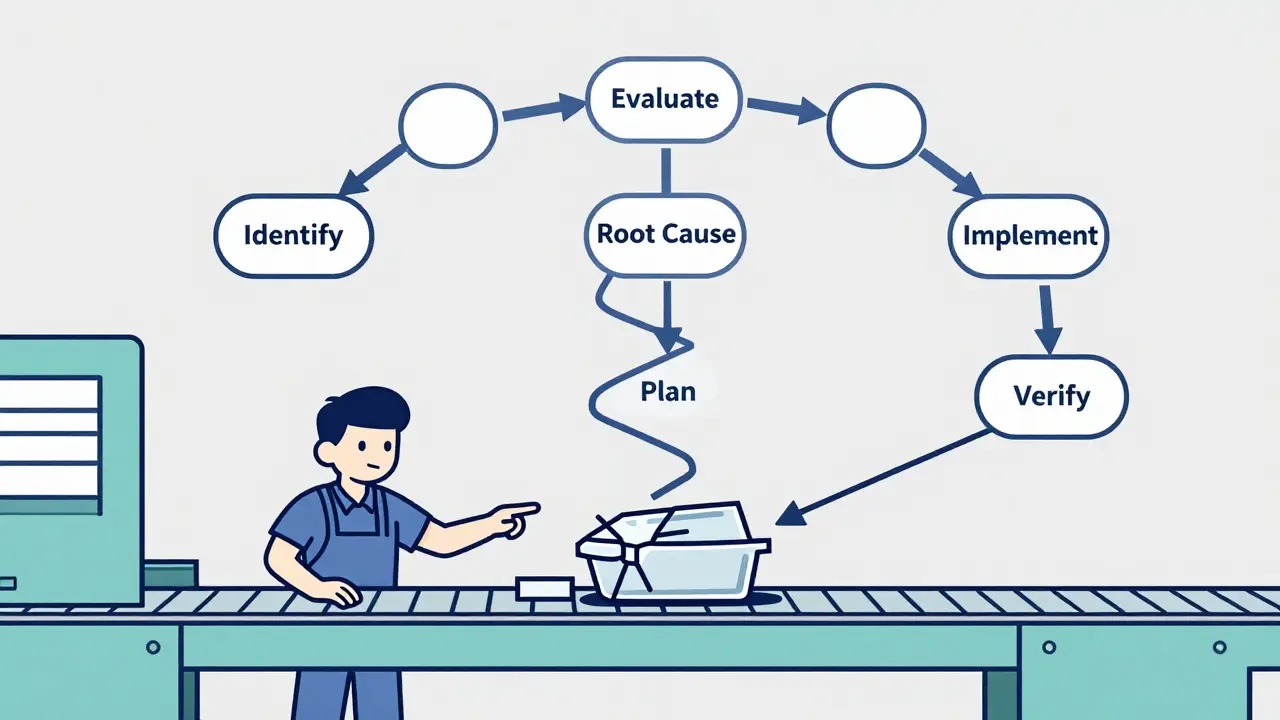
The Six Steps of a Real Corrective Action
A proper corrective action doesn’t happen in a day. It follows a clear, six-step process that turns a problem into a permanent fix.
- Identify the problem - It starts with data. A quality inspector notices 12 out of 500 units have cracked housings. A machine sensor flags abnormal vibration. A customer returns a batch with inconsistent labeling. The trigger isn’t a complaint-it’s a measurable deviation from the standard.
- Evaluate and prioritize - Not all defects are equal. A mislabeled box might be a nuisance. A contaminated drug vial is a crisis. Manufacturers use risk assessments to decide which issues demand full CAPA. High-risk problems get immediate attention. Low-risk ones might just get logged and monitored.
- Find the root cause - This is where most companies fail. Too many teams stop at the surface: “The machine was out of alignment.” But why was it out of alignment? Was the calibration schedule missed? Was the technician trained? Was the tool worn and not replaced? Tools like the 5 Whys and Fishbone diagrams force teams to dig past symptoms. One medical device maker found their packaging defect wasn’t due to the sealer-it was because a new operator wasn’t trained to check the seal temperature after a shift change.
- Plan the fix - Once you know the real cause, you design a solution. This isn’t a vague idea. It’s a detailed plan: “Replace the manual calibration log with an automated system that locks the machine if calibration is overdue. Assign QA lead Maria Chen to verify compliance weekly. Complete by March 15.” Every action has a name, a deadline, and a way to prove it happened.
- Implement and document - The fix goes live. Training records are updated. Machine settings are changed. New inspection checklists are printed. Every step is recorded. No email. No sticky note. It’s in the system. Auditors don’t believe what you say-they believe what you’ve written down.
- Verify effectiveness - The final step is the most ignored. Did the fix actually work? After implementation, manufacturers track the defect rate for at least three full production cycles. If the defect dropped from 2.8% to 0.4%, that’s proof. If it stayed at 2.5%, the fix failed. And you go back to step three.
Why Most Corrective Actions Fail
According to FDA inspection data from 2022, 43% of warning letters cited weak CAPA systems. The biggest reason? People fix the symptom, not the cause.
One automotive supplier kept seeing bent shafts on their assembly line. Their first fix? Add a visual inspection station. The defect rate dropped slightly-but not enough. After digging deeper, they found the bending happened because a new supplier had changed the raw material’s hardness without telling them. The real fix? A new material certification process with incoming inspection specs. That cut the defect rate by 90%.
Another common failure is poor verification. A company says, “We fixed it,” but never checks if the fix stuck. They might run a few test units and call it good. But real verification needs statistical proof-usually at least 30 samples tested under normal production conditions. Without that, you’re guessing.
And then there’s the paperwork problem. One Reddit user, ‘FactoryQA’, said their CAPA for a single issue generated 47 pages of documents. That’s not control-it’s paralysis. The best systems cut the noise. They use digital tools that auto-populate forms from machine data, link inspection results to root cause analyses, and flag delays before they become disasters.
Corrective Action vs. Correction vs. Preventive Action
People mix these up all the time. Here’s the difference:
- Correction is a quick patch. Stop the line. Throw out the bad parts. Adjust the torque setting. It fixes the immediate problem, but doesn’t stop it from happening again.
- Corrective action is the deep fix. It finds why the machine was misadjusted in the first place and changes the system so it won’t happen again.
- Preventive action is forward-looking. It’s not about something that already broke. It’s about spotting a pattern-like 15 near-misses in the last month-and changing the process before the next failure occurs.
Confusing these three is the #1 reason quality systems collapse. In fact, 68% of manufacturing failures come from treating a correction like a corrective action. A pharmaceutical company once fixed a labeling error by retraining staff (a correction). They didn’t realize the label design itself was confusing. Later, the same error happened again-this time with a life-threatening dosage mix-up. That’s when they finally did the real corrective action: redesigned the label based on human factors testing.
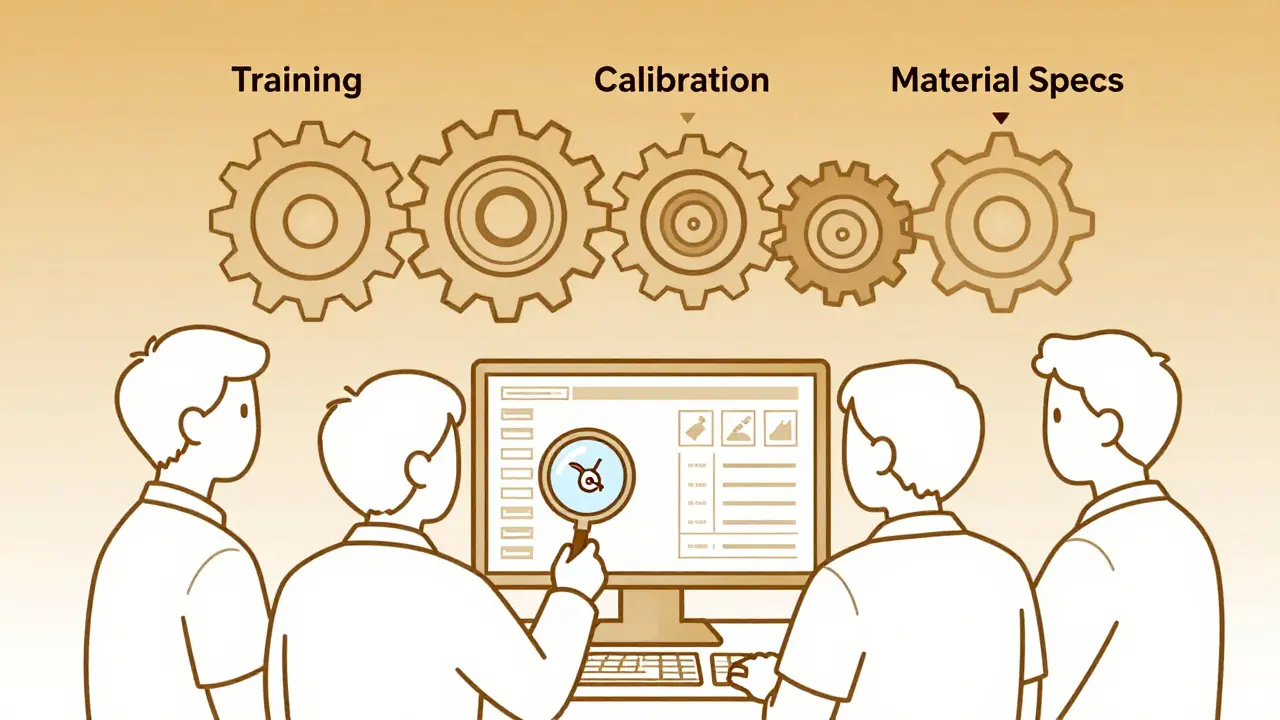
How Digital Tools Are Changing the Game
Manually tracking CAPAs in Excel or paper logs is a relic. Today’s best manufacturers use integrated quality management software that connects machines, sensors, and people.
One New Zealand-based medical device maker installed IoT sensors on their injection molding machines. When a mold temperature drifted outside tolerance, the system didn’t just alert the operator-it auto-generated a CAPA ticket, pulled historical data from the last 50 similar events, and suggested the most likely root cause based on AI patterns. The investigation time dropped from 10 hours to 4.8 hours. Accuracy improved by 37%.
These systems also make verification easier. Instead of manually collecting 30 samples and checking them one by one, the software pulls real-time defect data from the production line. If the defect rate stays below 0.5% for three consecutive batches, the CAPA closes automatically.
The FDA is pushing this shift. Their 2023 Digital Health Innovation Plan encourages blockchain-backed audit trails and electronic CAPA systems. Already, 41% of medical device firms are testing these tools. By 2027, Gartner predicts 65% of manufacturers will use predictive CAPA-systems that spot trends before defects even occur.
What Success Looks Like
Companies that do CAPA right see real results:
- 37% less downtime from quality issues
- 28% higher customer satisfaction scores
- 19% lower costs from reduced scrap and rework
- 27% fewer product recalls and field actions
One company in the Midwest cut their defect rate from 2.8% to 0.4% in 18 months-not by hiring more inspectors, but by fixing one root cause: inconsistent tooling maintenance. They assigned one person to own every tool’s calibration schedule. That’s it. No new machines. No new software. Just accountability.
Success isn’t about having the fanciest system. It’s about having a culture where people feel safe saying, “I don’t know why this happened,” and where leaders reward digging deep instead of rushing to fix the symptom.
13 Comments
This is the kind of post that makes me feel hopeful about manufacturing again. Too often we just band-aid things and move on, but real change happens when we slow down and ask why. Thanks for laying it out so clearly.
The distinction between correction and corrective action is everything. I've seen entire departments collapse because leadership confused the two. It's not about fixing the symptom-it's about redesigning the system that allowed the symptom to exist in the first place.
Frankly, the entire CAPA framework is a glorified compliance theater unless you're leveraging predictive analytics and closed-loop feedback systems. The 5 Whys? Cute. But without Bayesian inference modeling on historical defect clusters, you're just doing root cause bingo. And let's not pretend manual verification with 30 samples is statistically valid-power analysis is non-negotiable in any real quality system.
People think digital tools are the answer. They're not. The real problem is that no one in management has the intellectual honesty to admit they don't understand the process. You can automate everything, but if your QA lead is just checking boxes because they're afraid of getting fired, you're still going to have recalls.
I've trained teams in 12 countries on this. The biggest cultural barrier isn't the process-it's fear. People don't report problems because they think they'll be blamed. The moment leadership responds to a CAPA with curiosity instead of anger, everything changes. That's the real magic.
The most overlooked part of step six is the time horizon. Three production cycles isn't enough for low-volume, high-complexity systems. In aerospace, we track for six months minimum. One defect recurrence after 90 days is still a failure. You can't rush validation when lives are on the line.
I love how you mentioned the paperwork problem. My team used to spend 60 hours a week on CAPA documentation. We switched to a tool that auto-generates from machine logs and now we have time to actually fix things. Sometimes the best innovation is just cutting the noise.
This gave me chills. In India, we're just starting to adopt these practices, and it's amazing to see how simple changes-like assigning one person to own tool calibration-can make such a huge difference. Thank you for sharing this!
lol i read this whole thing and still dont know what a fishbone diagram is but i get the point. stop fixing the symptom. fix the system. got it.
So let me get this straight. You're telling me the solution to 43% of FDA warning letters is... not being lazy? Wow. Groundbreaking. I'm sure the executives who ignore CAPA because it 'slows down production' are just kicking themselves right now.
The example of the automotive supplier is textbook. I’ve seen this exact scenario play out in three different plants. The first fix always involves adding a step. The real fix always involves changing the supplier contract or material spec. The lesson? The problem is rarely on your line-it’s upstream.
Verification isn't optional-it's the only step that proves the system works. I've audited CAPAs where the 'evidence' was a single screenshot of a dashboard. That's not verification. That's wishful thinking. Real verification requires statistical control charts, control limits, and a documented baseline. Anything less is just noise.
The 68% statistic is misleading. Most failures occur because corrective actions are assigned to overworked engineers who have no authority to change processes. The system isn't broken-the power structure is.