The pharmaceutical industry runs on a delicate balance between innovation and competition. At the heart of this balance in the United States is the 180-day exclusivity, which is a statutory incentive that grants the first generic manufacturer to challenge a brand-name patent exclusive marketing rights for six months. This provision, born from the Hatch-Waxman Amendments, was designed to encourage generic companies to take on costly patent litigation by offering them a temporary monopoly. However, there is a major loophole that complicates this reward: authorized generics.
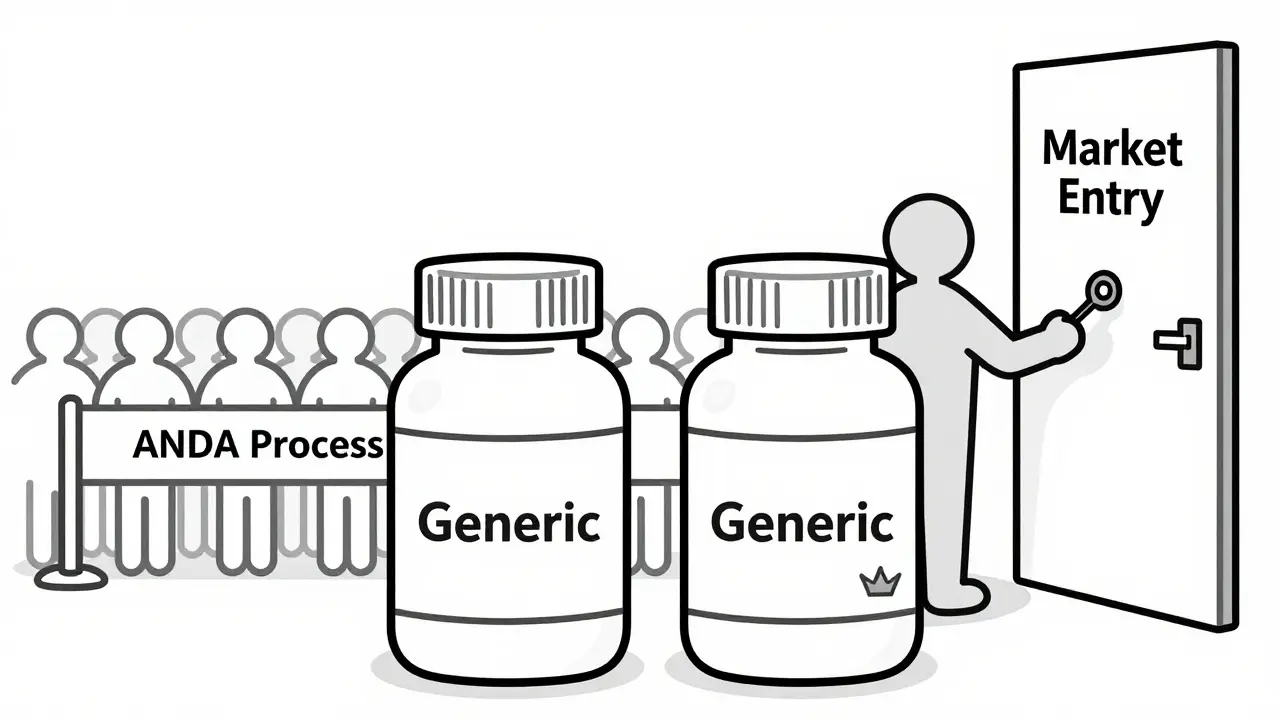
An authorized generic is the exact same brand-name drug sold without the brand name on the label, manufactured by the original brand company or its licensee. These products do not need to go through the rigorous Abbreviated New Drug Application (ANDA) process that independent generics must follow. Instead, they enter the market immediately alongside the first generic entrant. This creates a complex legal and competitive landscape where the intended "bounty" for challenging patents is often shared-or stolen-by the very brand-name companies those generics were trying to displace.
How the 180-Day Exclusivity Clock Starts
To understand why authorized generics are such a disruptive force, you first need to know how the exclusivity period works. Under 21 U.S.C. § 355(j)(5)(B)(iv), the law defines the 180-day period as ending on the day before an application submitted by an applicant other than the first applicant is approved. In simpler terms, no other generic competitor can get FDA approval during these 180 days.
The clock starts ticking when one of two things happens:
- Commercial Marketing: The first generic applicant actually ships the product to customers.
- Court Decision: A court rules that the brand-name patent is invalid, unenforceable, or not infringed.
Whichever event occurs first triggers the start date. This mechanism is crucial because it means the exclusivity is tied to action, not just approval. If a generic company wins the legal battle but delays launching the product to build up inventory, they are burning through their exclusive window without making sales. The FDA clarified in its 2017 guidance that "first commercial marketing" requires both final approval and actual shipment. Mistakes here are common; the FDA reports that about 28% of first generic applicants between 2018 and 2022 lost some portion of their exclusivity due to procedural errors in triggering the period correctly.
The Rise of Authorized Generics
While the first generic is fighting for its 180-day head start, the brand-name manufacturer has a secret weapon. They can launch an authorized generic. Because this product is essentially the brand-name drug with a different label, it does not face the same regulatory hurdles. It doesn't need to prove bioequivalence again, nor does it need to wait for the 180-day exclusivity to expire.
This strategy has become increasingly common. Data cited in congressional testimony shows that between 2005 and 2015, brand-name manufacturers launched authorized generics in approximately 60% of cases where 180-day exclusivity was granted. For the generic challenger, this is a nightmare scenario. Instead of enjoying a near-monopoly for six months, they immediately share the market with the original innovator.
The financial impact is stark. Research indicates that when an authorized generic enters during the exclusivity period, the first generic's market share drops from an average of 80% to just 50%. This reduces potential revenue by 30% to 50%. In high-stakes cases, like Teva Pharmaceuticals' litigation against Eli Lilly over Humalog, Teva estimated losing $287 million in revenue specifically because Lilly launched an authorized generic during Teva's exclusive window.
| Feature | First Generic (Paragraph IV) | Authorized Generic |
|---|---|---|
| Regulatory Pathway | Abbreviated New Drug Application (ANDA) | New Drug Application (NDA) derivative |
| Patent Challenge Required? | Yes (Paragraph IV Certification) | No |
| Market Entry Timing | After FDA approval + litigation resolution | Immediately upon generic entry |
| Exclusivity Protection | 180 days from other generics | None (can enter anytime) |
| Average Market Share (during exclusivity) | ~50-80% (if no auth generic) | Competes directly with first generic |
Legal Battles and Antitrust Concerns
The tension between these two forces has led to significant legal controversy. The Federal Trade Commission (FTC) views the use of authorized generics as a potential antitrust violation in many contexts. Between 2010 and 2022, the FTC filed 15 antitrust lawsuits against brand-name manufacturers for allegedly using authorized generics to improperly delay or dampen generic competition.
The core argument is that while authorized generics technically lower prices compared to the brand name, they undermine the Hatch-Waxman framework's goal of encouraging robust generic competition. By fragmenting the market early, brand companies prevent the first generic from capturing enough profit to justify the risk of patent litigation. This creates a "chilling effect," discouraging smaller generic companies from taking on expensive legal battles.
Industry professionals note that the threat of authorized generics has made Paragraph IV challenges far less attractive for smaller firms. Challenging a single drug patent can cost $2 to $5 million in legal fees alone. If the resulting market share is slashed by half due to an authorized generic, the return on investment becomes questionable. As a result, larger generic manufacturers now routinely negotiate contractual provisions with brand-name companies to delay authorized generic entry as part of patent settlement agreements. In 2022, 78% of first generic applicants included such clauses in their settlements.

Legislative Efforts to Close the Loophole
Recognizing this imbalance, lawmakers have repeatedly tried to fix the system. The Preserve Access to Affordable Generics and Biosimilars Act, introduced in various forms since 2009 and reintroduced in the 118th Congress, aims to prohibit brand-name manufacturers from launching authorized generics during the 180-day exclusivity period.
FDA Commissioner Robert Califf testified in March 2023 that the agency supports clarifying the law to prevent this practice. The FTC’s 2022 Pharmaceutical Report also recommended amending the Hatch-Waxman Act to explicitly exclude authorized generics from the exclusivity period. Analysts project that if this legislation passes, the value of successful Paragraph IV challenges could increase by $150 to $250 million per drug. This would likely lead to a 20-25% increase in patent challenges, accelerating patient access to cheaper medicines.
However, brand-name pharmaceutical companies argue against this change. They claim that authorized generics benefit consumers by lowering prices immediately upon generic entry. A 2021 RAND Corporation study supported this view, showing that prices are 15-25% lower when authorized generics compete with the first generic entrant compared to scenarios with only one generic competitor. The debate highlights the fundamental tension in pharmaceutical policy: balancing immediate price reductions with long-term incentives for generic innovation.
Strategic Considerations for Stakeholders
For generic manufacturers, the current environment requires sophisticated strategy. You cannot simply file an ANDA and hope for the best. Companies must establish cross-functional teams involving regulatory affairs, legal, and commercial personnel at least six months before anticipated approval. These teams work to optimize the timing of the "trigger" event to ensure maximum market capture.
Key pitfalls to avoid include:
- Miscalculating the trigger date: Starting the clock too early by shipping sample units for testing rather than commercial sale.
- Failing to maintain continuous marketing: Interruptions in supply can forfeit exclusivity.
- Underestimating authorized generic risk: Not factoring in the likelihood of brand entry into financial projections.
For policymakers and healthcare providers, understanding this dynamic is crucial. The 180-day exclusivity provision has accelerated patient access to generics by an average of 3.2 years compared to the pre-Hatch-Waxman era, resulting in $2.2 trillion in healthcare savings since 1984. But the effectiveness of this incentive is diminishing. The average time from first generic approval to multiple generic competition has decreased from 28 months in 2000 to just 9 months in 2022, partly due to these aggressive authorized generic strategies.
What is the difference between a generic drug and an authorized generic?
A generic drug is produced by a different manufacturer than the brand-name company and must go through the FDA's ANDA process to prove it is bioequivalent to the brand. An authorized generic is the exact same product as the brand-name drug, made by the brand manufacturer or its licensee, but sold under a different label. It does not need to undergo the full generic approval process.
How does the 180-day exclusivity period begin?
The period begins on the earliest of two dates: the date the first generic applicant commences commercial marketing (ships the product to customers) or the date a court finds the relevant patent invalid, unenforceable, or not infringed. It does not start simply upon FDA approval.
Can an authorized generic be launched during the 180-day exclusivity period?
Yes. Current law allows brand-name manufacturers to launch authorized generics at any time, including during the 180-day exclusivity period granted to the first generic entrant. This is a major point of legal and political controversy, with proposed legislation aiming to ban this practice.
Why do brand-name companies launch authorized generics?
Brand companies launch authorized generics to compete directly with the first generic entrant, thereby reducing the generic's market share and profitability. This strategy helps the brand retain more revenue than if they allowed the generic to have a complete monopoly for 180 days. It also allows them to offer lower prices than the brand name, potentially keeping patients in their ecosystem.
What is a Paragraph IV certification?
A Paragraph IV certification is a statement filed by a generic manufacturer in their ANDA asserting that the patents listed in the FDA's Orange Book for the brand-name drug are either invalid, unenforceable, or will not be infringed by the generic product. Filing this certification is a prerequisite for receiving 180-day exclusivity if successful.
Has legislation been proposed to stop authorized generics during exclusivity?
Yes. The Preserve Access to Affordable Generics and Biosimilars Act has been introduced multiple times, most recently in the 118th Congress. It seeks to prohibit brand-name manufacturers from marketing authorized generics during the 180-day exclusivity period. While supported by the FDA and FTC, it has not yet passed into law.